




Esclerose tuberosa
Autores: Davi Ferreira Soares, Dr. Igor Zamilute.
Dr. Igor Zamilute
Orientador: Prof. Dr. Fabiano Reis
História Clínica
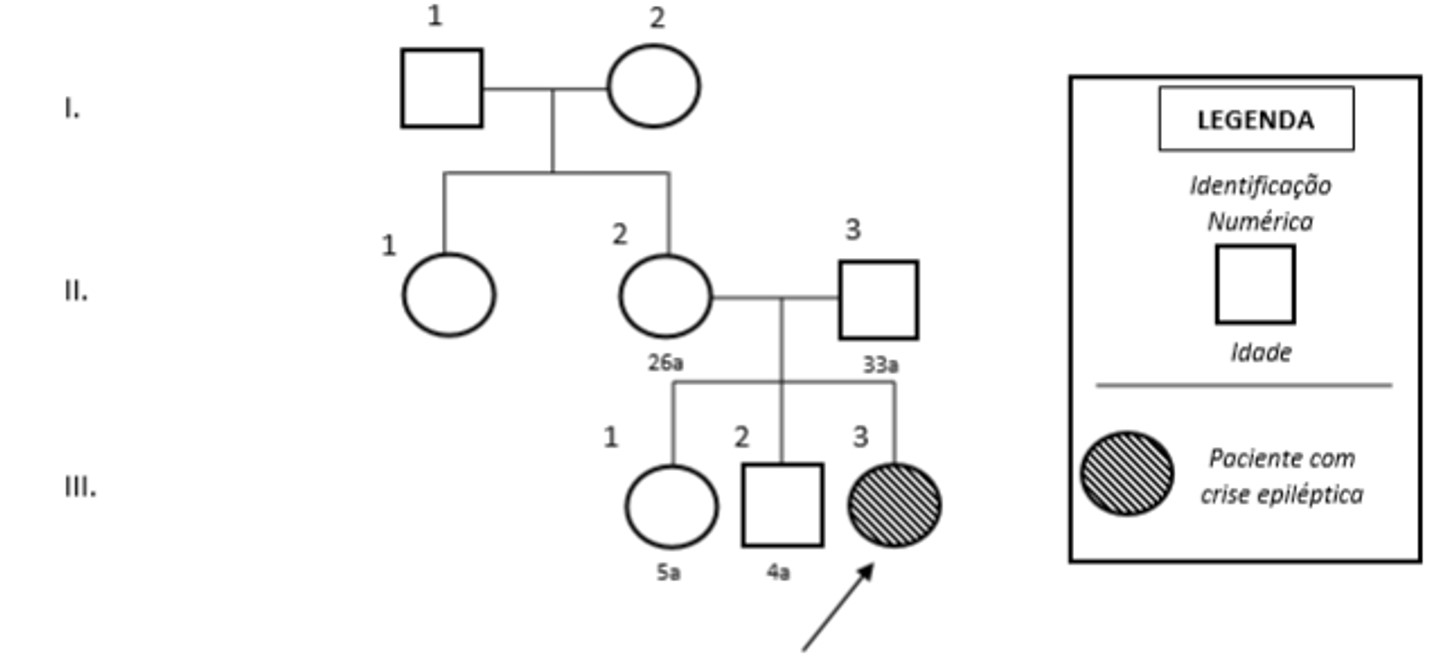
Paciente sexo feminino, 2 anos de idade. Encaminhado por outro serviço de saúde para o pronto-socorro da pediatria por conta de quadro inicial de febre, vômitos e infecção do trato urinário (ITU) em resolução, evoluindo para episódio convulsivo tônico clônico generalizado (TCG) e rebaixamento do nível de consciência. Mãe nega intercorrências durante gestação (G3C3A0), mas aos 2 meses de idade notou que paciente apresentava hipotonia, dificuldade para andar, falar e alteração da movimentação ocular. Na mesma época, manifestou primeiro episódio TCG e novamente com 5 meses, ficando desde então sem crise. Nega história familiar de crises epilépticas e consanguinidade (ver heredograma).
Exame Físico
PA: 110 x 60 mmHg FC= 136 bpm Temperatura: 37 0C
Mal estado geral, descorada +1/+4, anictérica, afebril e acianótica. Inconsciente. Movimentos TCG em extremidades. Atraso do desenvolvimento neuropsicomotor. Midríase (pouco responsiva bilateralmente). Sem massas e visceromegalias abdominais.
Exames Complementares
ECG: Sem alterações.
EEG: Traçado revela distúrbio cerebral inespecífico na região centro-temporal direita e atividade epileptiforme pouco frequente na região têmporo-occipital direita.
TC –Crânio: Nódulos calcificados no pedúnculo cerebelar médio esquerdo, cabeça do núcleo caudado direito, subependimários nos ventrículos laterais e na substância branca dos centros semi-ovais. Lesões hiperdensas nodulares sem impregnação no córtex frontal direito e esquerdo. Áreas hipodensas córtico/subcorticais temporal e parietal direita, frontal e parietal esquerda (compatíveis com túberes). Calcificações córtico-subcorticais, representativas de calcificações em túberes.
Diagnóstico
Esclerose Tuberosa
Imagens
Paciente já com 19 anos, realizou exames de imagem para acompanhamento da esclerose tuberosa
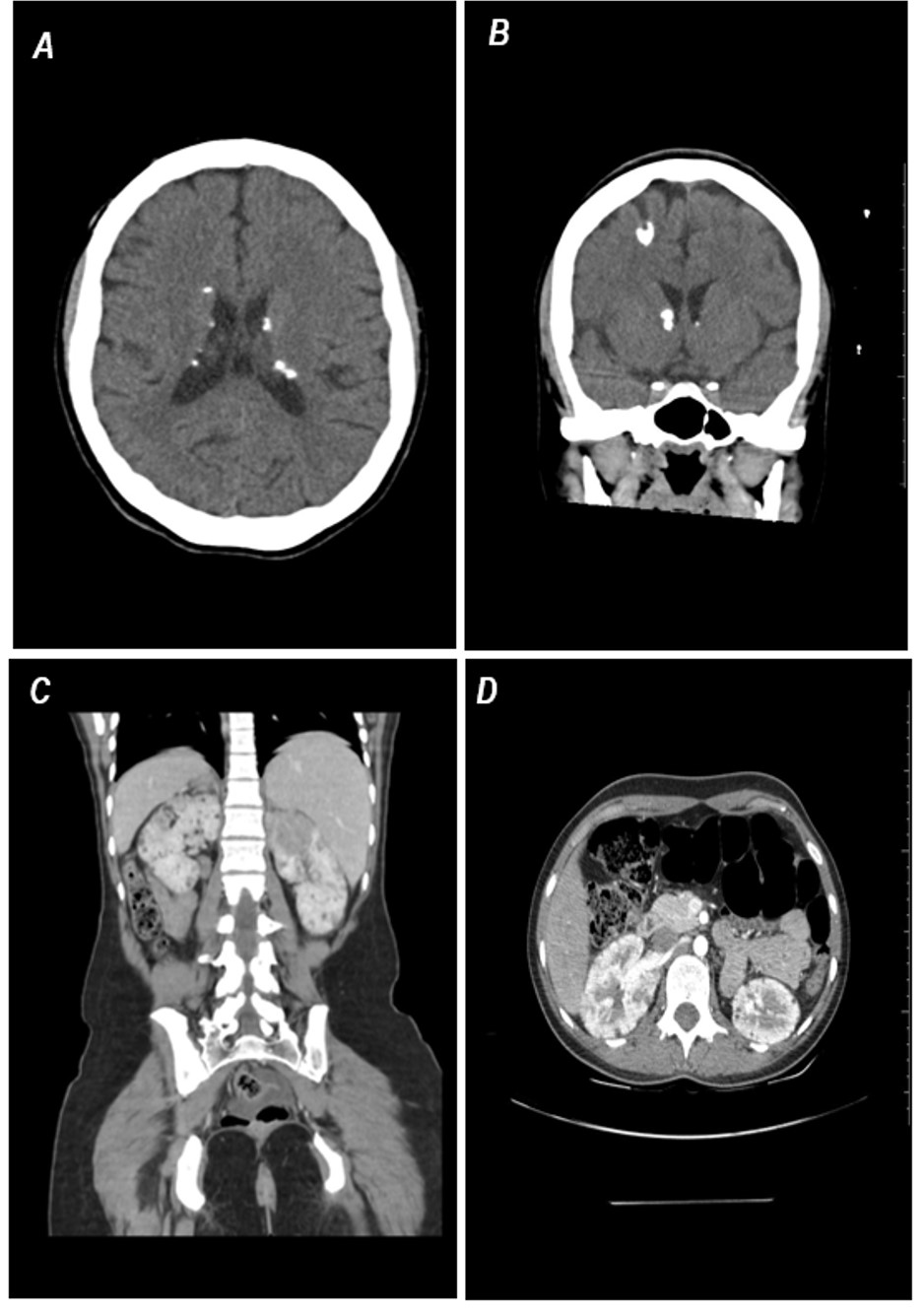
Tomografia de crânio em cortes axial (A) e coronal (B), evidenciando múltiplas calcificações grosseiras em topografia subependimárias, ao longo dos sulcos tálamo-caudados, compatíveis com nódulos subependimários calcificados.Nota-se ainda na alta convexidade frontal direita, área de distorção e expansão giral, de alta densidade, que representa túber córtico-subcortical calcificado.
Tomografia da abdome pós-contraste em cortes coronal (C) e axial (D), evidenciando formações nodulares hipodensas, com áreas com atenuação de gordura, em ambos os rins, compatível com múltiplas angiomiolimpomas. Destaque para hipodensidade evidenciada no terço superior do rim direito, contendo diminuto foco de gordura intralesional, representando angiomiolipoma pobre em gordura.
Discussão
A esclerose tuberosa (ET) é uma doença autossômica dominante e pleiotrópica, gerando múltiplas manifestações principalmente em sistema nervoso central, rim, pulmão, coração e pele. Alguns estudos classificam-na como facomatose (relacionada à origem ectodérmica) tal como neurofibromatose I e II e doença de Von Hippel-Lindau. Os genes responsáveis pela ET são TSC1 (cromossomo 9) e TSC2 (cromossomo 16), sendo que 60% a 70% dos casos são esporádicos, e mutações do TSC1 são duas vezes mais frequentes em casos familiares.
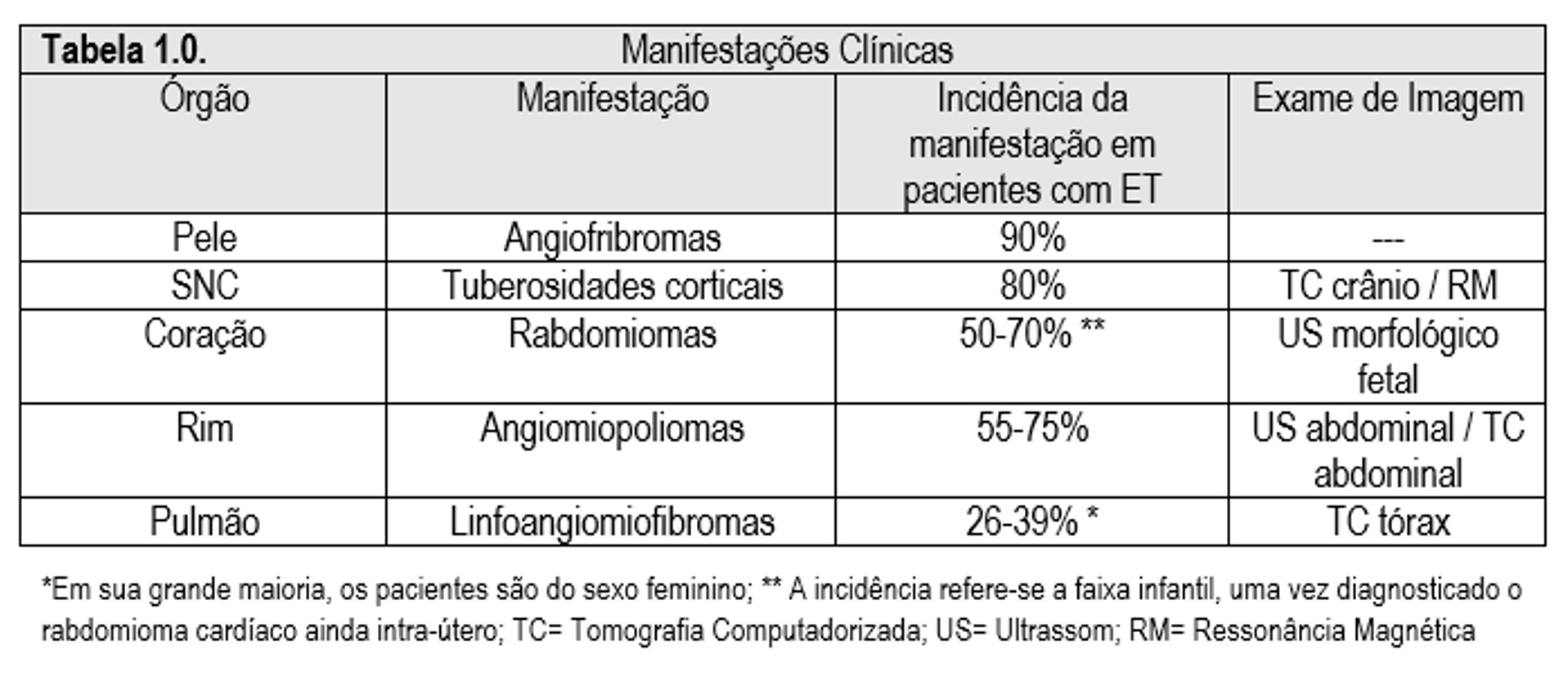
Em suma, ambos os genes estão envolvidos na via mTOR, ligada às cascatas de crescimento celular, explicando a fisiopatologia da ET, uma vez que células ectodérmicas e mesodérmicas sofrem hiperplasia e alteração em sua migração celular, levando ao aparecimento de tumores cerebrais, renais, cardíacos (no coração, os rabdomiomas são característicos) e pulmonares, este sendo característico em mulheres. O modelo de genético de Knudson infere a necessidade de inativação de ambos os alelos de TSC1 ou TSC2 para manifestação das lesões típicas da ET. Estas manifestações estão resumidas na Tabela 1.0.
Sobre os aspectos clínicos, o paciente geralmente procura serviço de saúde por conta das lesões de pele, principalmente os angiofibromas faciais e manifestações neurológicas, incluindo crises epilépticas, autismo, transtornos psiquiátricos e deficiência intelectual.
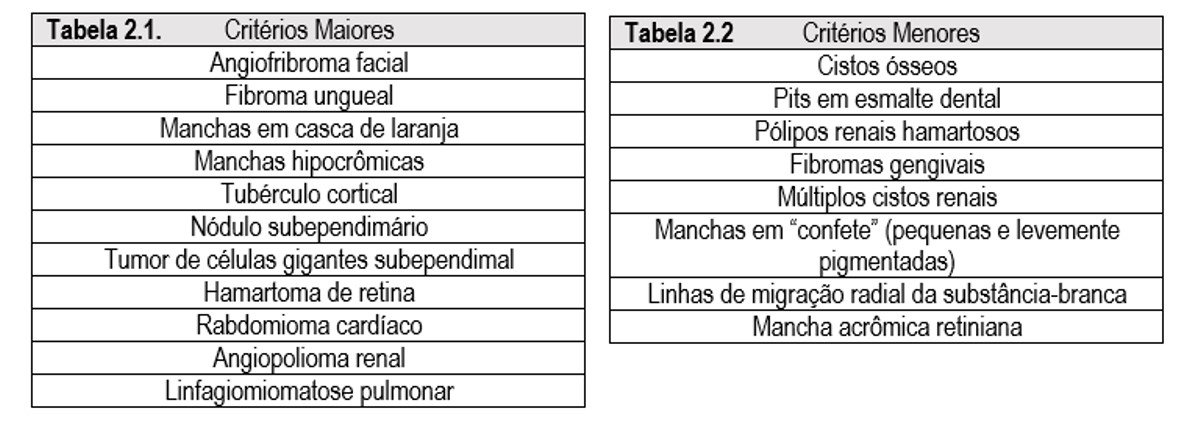
O diagnóstico é feito segundo critérios maiores e menores (Tabela 2.1 e 2.2, respectivamente), sendo necessário dois maiores ou um maior associado à dois menores. Ainda não há um guideline específico para acompanhamento da doença, entretanto orienta-se que sejam realizados exames de imagem abdominal e cerebral a cada três anos. Em pacientes menores de 21 anos, sugere-se RM cerebral anual, e a cada 2-3 anos após 21 anos para monitorar o surgimento de astrocitoma subependimário de células gigantes, um tumor bastante relacionado à ET, e que, caracteristicamente, ocorre na região de um dos forames interventriculares (Monroe). No SNC, também podem ser observadas as bandas radiais, notadamente em estudos de RM, que são linhas que se estendem da superfície ependimária dos ventrículos em região ao córtex, e representam distúrbios de migração neuronal. A respeito das alterações comportamentais, manifestações de autismo ou distúrbios psicopatológicos são necessários serem avaliados pelo serviço psiquiátrico para controlar, por exemplo, impulsividade e agressão, ajudando no desenvolvimento cognitivo. Pacientes com linfoangiomiomatose necessitam do monitoramento pulmonar a depender da sintomatologia. Por sua vez, o eletroencefalograma não constitui uma ferramenta para diagnóstico, entretanto é um auxílio importante para avaliar atividade cerebral e, na presença de epilepsia, identificar o foco epileptogênico. O acompanhamento dermatológico é útil, visto que angiofibromas faciais podem ser desfigurantes, necessitando de laser ou remoção cirúrgica.
Por fim, o aconselhamento genético é necessário frente a planejamento familiar, pois a ET é uma desordem autossômica dominante, implicando na probabilidade de 50% da próxima criança ser afetada. Ainda não há tratamento, embora estudos visando inibir mTOR mostram regressão tumoral em ratos, principalmente os subependimários.
Referências
1. Crino, P. B., Nathanson, K. L., Henske, E. P. The tuberous sclerosis complex. New Eng. J. Med. 355: 1345-1356, 2006
2. Curatolo, P., Bombardieri, R., Jozwiak, S. Tuberous sclerosis. Lancet 372: 657-668, 2008.
3. Dabora, S. L., Jozwiak, S., Franz, D. N., Roberts, P. S., Nieto, A., Chung, J., Choy, Y.-S., Reeve, M. P., Thiele, E., Egelhoff, J. C., Kasprzyk-Obara, J., Domanska-Pakiela, D., Kwiatkowski, D. J. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 68: 64-80, 2001.
4. Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 13: 624–28, 1998.
5. Hyman MH, Whittemore VH. National Institutes of Health consensus conference: tuberous sclerosis complex. Arch Neurol 57:662-5, 2000.
6. Yvonne Baron, A. James Barkovich. MR Imaging of Tuberous Sclerosis in Neonates and Young Infants. AJNR Am J Neuroradiol 20: 907-916, 1999.
Autores:
Davi Ferreira Soares: Aluno do 4o ano do curso de graduação em Medicina da Faculdade de Ciências Médicas –FCM / Unicamp, SP, Brasil.
Dr. Igor Zamilute: Médico residente (R3) do Departamento de Radiologia da Faculdade de Ciências Médicas –FCM / Unicamp, SP, Brasil.
Orientador:
Prof.Dr. Fabiano Reis: Professor Doutor do Departamento de Radiologia da Faculdade de Ciências Médicas –FCM /Unicamp, SP, Brasil.



