




Síndrome de Sturge Weber
Autor: Guilherme Wertheimer
Orientador: Prof. Dr. Fabiano Reis
História Clínica
Paciente pardo do sexo masculino, com 6 meses de idade comparece ao Pronto-Atendimento com queixa de vômitos, hipoatividade e pausas respiratórias desde o dia anterior. Mãe relata que há 12 horas teve um episodio de vômito acompanhado de sudorese intensa, taquicardia e febre não aferida.
Em tempo se apresentou afebril, sem sinais de desidratação, com último episodio de vômito há 8 horas. Paciente submetido a triagem infecciosa com resultados negativos e segue para observação no PS evoluindo com episodio de hiporesponsividade, desvio ocular conjugado para esquerda e nistagmo por alguns segundos após 1 hora da chegada. O quadro se repete 5x em 5minutos de observação e é notada ascensão da temperatura para 37,6˚C e queda da saturação O2 para 91%. Foi prescrito diazepan 0,2mg/kg com uso de traqueia O2 levando a melhora dos sintomas. Paciente segue para internação na enfermaria para investigação de crises convulsivas a esclarecer.
Na internação paciente apresenta crises semelhantes com abdução em membro inferior direito, movimentos mastigatórios e movimentação clônicas em membros superiores, sendo solicitados tomografia computadorizada (TC) e posteriormente ressonância magnética do crânio (RM).
Achados Clínicos
Paciente em bom estado geral, corado, hidratado, acianótico, anictérico, afebril (36˚C) e eupneico. Sem linfonodos palpáveis nas cadeias cervical e supraclaviculares. Fontanela anterior normotensa sem abaulamento, tempo de enchimento capilar <2’’ e sem edema de membros inferiores.
-Face: hemangioma plano em hemiface esquerda
-Torácico: MV presente bilateralmente sem ruídos adventícios e sinais de desconforto respiratório. Duas bulhas normofonéticas e rítmicas sem sopros. Frequência respiratória de 32 irpm e cardíaca de 120bpm. Saturação O2 98%.
-Abdômen: globoso e flácido com RHA presentes. Ausência de massas e visceromegalia com descompressão brusca negativa.
-Neurológico: reflexos simétricos, paralisia flácida a direita com tônus mantido à esquerda. Preferência por movimentação do lado esquerdo ao direito. Pupilas isocóricas e fotorreagentes.
Hipóteses Diagnósticas:
-Estado de mal não convulsivo
-Síndrome de Sturge-Weber
Exames por imagem
Tomografia computorizada de crânio: SEM E COM CONTRASTE
-Vasos do polígono de Willis com morfologia normal.
-Redução volumétrica da região parieto-temporo-occipital esquerda, calcificação linear cortical (de aspecto giriforme), realce de vasos no espaço subaracnóide, há drenagem para o sistema venoso subependimário no átrio ventricular lateral, onde há proeminência do plexo coroide e para veias da convexidade cerebral e seio reto.
Ressonância magnética do crânio: obtida com técnicas de SE, FSE, IR e angio-ressonância 3D/GRE, com imagens pesadas em T1 e T2 e aquisição multiplanar, antes e após a administração IV de contraste paramagnético (gadolínio), que evidencia:
-Alargamento dos sulcos cerebrais nas regiões temporo-parieto-occipital esquerdas, com leve atrofia do parênquima cerebral adjacente.
Alteração de sinal da substância branca subcortical nestas topografias (zonas de mielinização terminal), e, após a administração de contraste, impregnação de rede de capilares no espaço subaracnóide, em grande número, e que drenam no plexo coroide do ventrículo lateral esquerdo que está aumentado e para veias medulares que se dirigem aos seios reto e sagital superior. A angio-ressonância arterial demonstrou rede de vasos finos no espaço subaracnóide das regiões temporal e parieto-occipital esquerdas.
-HD: os achados descritos acima em associação com o quadro clínico de nevus vinhoso da face são compatíveis com angiomatose encefalotrigeminal (Síndrome de Sturge-Weber).
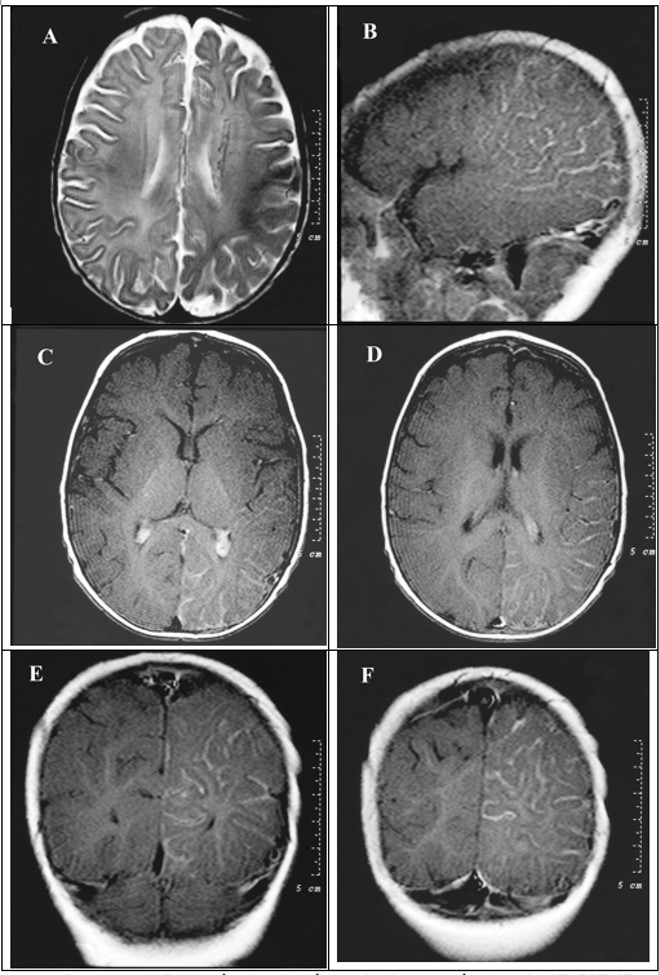
Ressonância magnética obtida com técnicas de SE, FSE, IR e angio-ressonância 3D/GRE, com imagens pesadas em T1 e T2 e aquisição multiplanar, antes e após a administração IV de contraste paramagnético (gadolínio). A: corte axial em T2 mostrando hipossinal da substância branca subcortical parieto-ocipital, provavelmente inferindo ocorrência de mielinização acelerada mas áreas acometidas pela patologia. B a F mostram áreas de realce leptomeningeo em regiões parieto-ocipitais à esquerda. B: corte sagital T1 pós contraste. C e D: cortes axiais T1 pós-contraste. E e F: cortes coronais T1 pós contraste.
Discussão
No caso em questão o paciente se apresentou ao PS com queixa de hipoatividade e paradas respiratórias, provavelmente associadas a crises convulsivas. No exame físico chamaram a atenção características como o lesão vinhosa de hemiface esquerda e hemiparesia flácida à direita, sendo requisitado exames de imagem para melhor investigação diagnóstica. A neuroimagem é essencial neste caso para o esclarecimento da etiologia das crises convulsivas e o diagnóstico da síndrome responsável pelas alterações observadas.
A síndrome de Sturge-Weber (SSW) ou angiomatose encefalotrigeminal, é uma síndrome neurocutânea rara e esporádica que acomete a microvasculatura venosa encefálica (1). Sua frequência estimada é entre 1:20.000 a 1:50.000 nascidos vivos nos Estados Unidos da América (2). A síndrome é causada por mutações somáticas em mosaico no gene GNAQ, envolvido na sinalização intracelular e cuja alteração, dependendo da fase do período embrionário, pode resultar em SSW ou manchas vinho do porto não sindrômicas (3). A SSW tem sinais marcantes como angiomatose da pele, olhos e leptomeninges, tipicamente se expressando como hemangiomas planos faciais na divisão oftálmica do nervo trigêmeo, também conhecidos como manchas vinho do porto (MVP), angiomatose leptomeningeal occipital ipsilateral, glaucoma e alterações vasculares oculares (4).
O quadro clínico da SSW é caracterizado por malformação dos capilares faciais com uma relação de 8% entre a presença das MVP e a síndrome. As MVP quando afetando toda a distribuição do ramo oftálmico do nervo trigêmeo é um fator preditivo considerável para distúrbios neurológicos e oculares associados (78%), enquanto o risco para esses distúrbios é de 26% quando apenas parte do ramo é acometido (5). A angiomatose intracraniana se apresenta de forma unilateral e posterior na maioria dos casos, não se relacionando com a distribuição da MVP e não existindo relação da extensão desta com a lesão cerebral (6). O curso clínico da síndrome é variável, mas normalmente apresenta problemas neurológicos progressivos como convulsões, hemiparesia , cefaleia, episódios semelhantes a AVCs, problemas comportamentais, retardo mental e defeito no campo visual. Os sintomas tendem a se estabilizar com o tempo (7).
Além das MVP, a epilepsia é frequentemente se a forma de apresentação da SSW. No caso relatado a busca por atendimento se deu por sinais gerados por crises convulsivas, hiporreatividade do pós-ictal e paradas respiratórias nos períodos de crise, sendo que convulsões normalmente se iniciam nos dois primeiros anos de vida (6), sendo que 75% dos casos no primeiro ano de vida, 86% até o segundo ano de vida e 95% até o quinto ano de vida (8). Vale ressaltar que 30% dos casos ocorrem durante episódios febris e ocorre maior susceptibilidade de convulsões febris em qualquer idade na maioria dos pacientes com SSW (6). As crises são normalmente focais motoras que podem generalizar e estado do mal epiléptico não é incomum associado com fraqueza prolongada em um lado corporal e novo déficit no campo visual, referidos como episódios semelhantes a AVC (9).
A SSW com componente intracraniano deve sempre ser suspeitado em pacientes com MVP, especialmente associados à sintomas neurológicos como no caso. A ressonância magnética especialmente imagens em T1 somada ao contraste gadolínio com sequência de susceptibilidade magnética é a modalidade de exame de imagem recomendada para demonstração dos achados característicos da SSW na fase inicial pré-sintomática, incluindo mudanças leptomeningianas com dilatação das veias transmedular e periventricular (10). O plexo coroide alterado com dilatação associado e aumento está presente em crianças mais velhas e adultos com dilatação dos vasos venosos de drenagem profunda na região cortical afetada (7). O uso das técnicas de FLAIR e FLAIR pós contraste pode aumentar a sensibilidade para detectar os vasos anômalos em leptomeninge (11).
Na apresentação inicial na infância pode haver pouca evidencia de atrofia, mas pode haver evolução para atrofia e calcificação que podem ser evidenciadas pela tomografia computadorizada (7). A idade ideal para o rastreamento de crianças com MVP sem acometimento neurológico com a RM é incerto, mas há evidência que após o 1o ano de vida se a RM é normal e não houver convulsões ou outros sintomas neurológicos o risco de haver envolvimento de SSW intracraniana é pequeno (12). Um estudo de coorte recente avaliou a extensão de hemangiomas leptomenigeanos evidenciados na RNM comparados com o quadro clínico de SSW, encontrando que o início das crises ocorreram mais cedo em indivíduos com envolvimento hemisférico de angiomatose (13). O acometimento bilateral intracraniano é observado em 15% dos pacientes e associado a maiores incidências de crises convulsivas, estas com início mais precoce e pior prognóstica (14). Nos casos em sua maioria há envolvimento predominante em um hemisfério (15) e nos casos o acometimento de lobo frontal bilateral parece que se mostrar um marcador importante de imagem do prognóstico de desenvolvimento e motor, enquanto acometimento bitemporal consiste em um fator de risco para características autistas (15).
Os sintomas de convulsões, hemiparesia e disfunção cognitivas podem ser correlacionados com imagens com as técnicas de difusão e perfusão (16). RNM funcional visual mostrou que podem a síndrome está associada com padrões de ativações occipitais com especulação de que a técnica pode ser útil em decisões de manejo cirúrgico (17). Técnicas recentes como o SPECT e FDG PET podem ser capazes de identificar mudanças que precedem sintomas clínicos, assim como atrofia e calcificação na TC e RM, pela anormalidade na perfusão cerebral e mudanças no metabolismo da glicose, respectivamente (18, 19).
O manejo da SSW envolve o envolvimento multidisciplinar como neurologista, oftalmologista e dermatologista. O reconhecimento precoce e controle das crises convulsivas de modo agressivo com uso de drogas anti epilépticas é recomendado por poder prevenir convulsões e episódios semelhantes ao AVC e portanto evitando lesão neurológica progressiva (1,7). Em pacientes com epilepsia refratária as cirurgias como a lesionectomia, calosotomia e hemisferectomia podem ser uma opção de tratamento (20).
O diagnóstico precoce e controle das comorbidades da SSW como glaucoma e epilepsia são essenciais para o prognostico favorável do paciente. A vigilância cuidadosa por neuroimagem para identificação de paciente com risco significativo de complicações e facilitar manejo sintomático agressivo e precoce é uma opção para melhorar a mortalidade e morbidade dos pacientes no futuro (7).
Referências:
1- Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies.Lymphat Res Biol 2007;5:257e64.
2- Thomas-Sohl KA, Vaslow DF, Maria BL. Sturge-Weber syndrome: a review. PediatrNeurol 2004;30:303-310.
3- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med 2013; 368:1971.
4- Bodensteiner J, Roach E. Sturge-Weber syndrome: introduction and overview. Mt. Freedom, NJ: The Sturge Weber Foundation;1999.
5-Ch’ng S, Tan S. Facial port-wine stains e clinical stratification and risks of neuro-ocular involvement. J PlastReconstrAesthetSurg 2008;61:889e93.
6- Pascual-Castroviejo I,Pascual-Pascual SI, Velazquez-Fragua R, Viano J. Sturge-Weber syndrome: study of 55patients. Can J NeurolSci 2008;35:301e7.
7- Sudarsanam A, Ardern-Holmes SL.Sturge-Weber syndrome: from the past to the present.Eur J Paediatr Neurol. 2014 May;18(3):257-66.
8- Sujansky E, Conradi S. Outcome of Sturge-Weber syndrome in 52 adults. Am J Med Genet 1995;57:35e45.
9- Maria BL, Neufeld JA, Rosainz LC, et al. Central nervous system structure and function in Sturge-Weber syndrome:evidence of neurologic and radiologic progression. J ChildNeurol 1998;13:606e18.
10-Hu J, Yu Y, Juhasz C, et al. MR susceptibility weighted imaging (SWI) complements conventional contrast enhanced T1weighted MRI in characterizing brain abnormalities ofSturge-Weber Syndrome. J MagnReson Imaging2008;28:300e7.
11- Griffiths PD, Coley SC, Romanowski CA, Hodgson T, Wilkinson ID. Contrast-enhanced fluid-attenuated inversionrecovery imaging for leptomeningeal disease in children.AJNR Am J Neuroradiol 2003;24:719e23.
12- Piram M, Lorette G, Sirinelli D, Herbreteau D, Giraudeau B, Maruani A. SturgeeWeber syndrome in patients with facial port-wine stain. PediatrDermatol 2012;29:32e7.
13- Fogarasi A, Loddenkemper T, Mellado C, et al. Sturge-Webersyndrome: clinical and radiological correlates in 86 patients.IdeggyogySz 2013;66:53e7.
14- Bebin EM, Gomez MR. Prognosis in Sturge-Weber disease: comparison of unihemispheric and bihemispheric involvement. J Child Neurol. 1988 Jul;3(3):181-4.
15- Alkonyi B, Chugani HT, Karia S, Behen ME, Juhasz C. Clinical outcomes in bilateral Sturge-Weber syndrome. Pediatr Neurol 2011;44:443-449.
16- Miao Y, Juhasz C, Wu J, et al. Clinical correlates of white matter blood flow perfusion changes in Sturge-Webersyndrome: a dynamic MR perfusion-weighted imaging study.AJNR Am J Neuroradiol 2011;32:1280e5.
17- Bernal B, Altman N. Visual functional magnetic resonance imaging in patients with SturgeeWeber syndrome. PediatrNeurol 2004;31:9e15.
18- Chugani HT, Mazziotta JC, Phelps ME.Sturge-Weber syndrome: a study of cerebral glucose utilization withpositron emission tomography. J Pediatr 1989;114:244e53.
19- Reid DE, Maria BL, Drane WE, Quisling RG, Hoang KB.Central nervous system perfusion and metabolism abnormalities inSturge-Weber syndrome. J Child Neurol 1997;12:218e22.
20- Arzimanoglou A, AicardiJ. The epilepsy of Sturge-Weber syndrome: clinical features and treatment in 23 patients. ActaNeurolScandSuppl 1992;140:18e22.
Autor: Guilherme Wertheimer - Aluno do 5o ano do curso de Graduação em Medicina da Faculdade de Ciências Médicas: FCM/UNICAMP
Orientador: Professor Doutor Fabiano Reis - Departamento de Radiologia da FCM/UNICAMP.

Possui graduação em Ciências Biológicas-Modalidade médica pela Universidade Federal de São Paulo (2007-2010) com ênfase em Neurociências. Graduação em Medicina na Universidade Estadual de Campinas (UNICAMP) (2012-2017) com cursos internacionais, trabalhos extracurriculares e de iniciação científica em Neuroimagem. Atualmente residente de Radiologia e Diagnóstico de Imagem na UNICAMP (2019-2022).



